Published Posters
Cryo-electron Microscopy of Vitreous Sections (CEMOVIS) Application for Model Organisms
Kunihiro Uryu1,* , Xiaowei Zhao2 , Momoko Shiozaki2 , and Zhiheng Yu1,2
1Electron Microscopy Shared Resource, HHMI Janelia Research Campus, Ashburn, VA, United States
2Cryo-Electron Microscopy Shared Resource, HHMI Janelia Research Campus, Ashburn, VA, United States
*Corresponding author
Cryo-ultramicrotomy, developed by Bernhard in 1965 [1], has long been regarded as the pinnacle of achievement for electron microscopists. This technique allows biological samples to be sliced into ultrathin sections and examined in a cryo-electron micro-scope, revealing the most intricate subcellular structures without chemical fixation or staining. The advent of vitrification [2,3] and high-pressure freezing (HPF) technology [4,5] provided reliable methods for preserving cellular structures, and the introduc-tion of diamond knife to cryo-ultramicrotomy [6] offering cryo-ultramicrotomists reassurance in consistency of the quality [7]. On one hand, advancements in HPF systems, the precision of electron microscopes, and digital imaging technology have sig-nificantly enhanced cryo-EM imaging capabilities. On the other hand, continuous improvements in ultramicrotome technology and their supporting environments have further optimized the technique. These enhancements include precise control of cutting temperatures, the use of ionized nitrogen to neutralize electron charging, and the development of tools such as diamond knives, the lash/EM grid manipulator, and a specialized voltage charging function to press sections onto EM grids [8]. Despite recent advancements, the contribution of cryo-electron microscopy of vitreous sections (CEMOVIS) in the field remains suboptimal. It is believed that the method remains technically challenging (e.g., the yield of high-quality micrographs is low) and cutting artifacts often deter researchers from further pursuing the technique [9].
In our efforts to overcome these challenges, we revisited and refined the method through a series of experiments. One of the focuses was on applying this technique to model organisms such as C. elegans, Drosophila, and mouse biopsy tissues. These tis-sues were subjected to high-pressure freezing followed by cryo-ultramicrotomy at temperatures ranging from -146°C to -160°C. Cutting conditions were tested between 50 and 200 nm section thicknesses with 50 to 200 um sample size. The ultramicrotome environment was optimized by reducing humidity and introducing remedies to reduce statistic charging. To address the retention of tissue integrity posed by animal models with cuticles and wax coverings, we tested various cryoprotectants, including bovine serum albumin, dextran, 1-hexadecene, sodium alginate, or specially formulated cryoprotectant cocktails with and without de-tergents. Our work is ongoing, but we are pleased to share the progress we’ve made [10].
Image ozaf048484

Figure 1. a) Cryo-microtome chamber with a lash and grid holder. Extra groundings were applied to reduce electron charging. b) A gold grid was used to collect ribbons of cryo-sections. Voltage charge increased the chance of retention of section in an organized manner. c,d,e) CEMOVIS sections from C elegans. A low magnification view of cross section (c), An intermediate magnification view of peripheral of the worm showing its preservation of the cuticle (d). A high-magnification view of a membrane structure, containing a fusing membrane vesicle, and electron dense granules (e).
Reference
- Bernhard W. Année biol. (1965) 4, 5-19. PMID: 14303978
- McDowall AW et al. Journal of Microscopy (1983) 131, 1–9. https://doi.org/10.1111/j.1365-2818.1983.tb04225.x
- Dubochet J et al. Q. Rev. Biophys. (1988) 21, 129-228. DOI: 10.1017/s0033583500004297
- Moor H in Cryotechniques in biological electron microscopy, Steinbrecht RA and Zierold K (Eds.), Berlin, Heidelberg: Springer; 1987. DOI:10. 1007/978-3-642-72815-0_8
- Studer D et al. Scanning Microsc. Suppl. (1989) 3, 253-68. PMID: 2694271
- Michel M et al. Journal of Microscopy (1992) 166, 43-56. https://doi.org/10.1111/j.1365-2818.1992.tb01506.x
- Al-Amoudi A et al. J. Struct. Biol. (2004) 148, 131-135. https://doi.org/10.1016/j.jsb.2004.03.010
- Studer D et al. Journal of Structural Biology (2014) 185, 125-128. https://doi.org/10.1016/j.jsb.2013.11.005
- Caspy I et al. Q. Rev. Biophys. (2025) 58, e6. DOI: https://doi.org/10.1017/S0033583525000010
- The authors acknowledge the funding from the HHMI support this study. We thank collegues from Janelia Research campus, esecially Drs. E Kuger and R Ikegami for scientific discussions and Mr. H Gnäegi in Diatome for technical support.
Time-Snapshot Volume Electron Microscopy Analysis of RNA Virus Replication Complex Assembly
Hong Zhan1,2,* , Adam Jochem1,2 , Mark Horswill1,2 , Johan den Boon1,2 , Pierre Gillotay3,4 , Kenneth Poss3,4 , and Paul Ahlquist1,2,5,*
1Rowe Center for Virology, Morgridge Institute for Research, Madison, WI, USA
2Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI, USA
3Department of Cell and Regenerative Biology, University of Wisconsin-Madison, Madison, WI, USA
4Morgridge Institute for Research, Madison, WI, USA
5McArdle Laboratory for Cancer Research, University of Wisconsin-Madison, Madison, WI, USA
*Corresponding author: [email protected], [email protected]
Recent advances in imaging technologies, from super-resolution light microscopy to volume electron microscopy (EM), have greatly expanded our ability to visualize the architecture of cells at multiple scales [1-5] . These methods now permit the study of structures ranging from individual proteins to entire cells or tissues, offering detailed insights into subcellular organization. Each approach faces challenges that can limit one or more aspects of cellular organization. Light microscopy can localize fluo-rescently tagged molecules but lacks ultrastructural resolution; conventional EM provides high-resolution details of subcellular ultrastructures but has limited scalability for capturing whole-cell volumes at nanometer resolution. Furthermore, chemical fix-ation can introduce artifacts, including morphological distortions.
A 3D EM technique, known as Volume EM, has emerged as a powerful tool for investigating cellular and tissue architecture at high resolution. In this approach, plastic-embedded sections are mounted on coverslips or grids and imaged by scanning electron microscopy (SEM) to build high-resolution, large-scale 3D datasets. In addition, the use of “epitope-friendly” plastic embedding materials, such as Lowicryl HM20 and LR White, permits immunolabeling of sections on glass coverslips. This workflow can be combined with correlative light and electron microscopy, often termed array tomography, to investigate specific proteins within their subcellular context [6-11].
Positive-strand [(+)] RNA viruses rearrange intracellular membranes to create virus-induced compartments or spherules, which serve as hubs for genome replication and virion assembly. These replication complexes undergo dynamic changes over the course of infection, involving interactions with host proteins and organelles essential for virus replication and survival. However, con-ventional transmission electron microscopy (TEM) captures only a thin slice of the cell, making it difficult to reconstruct the full organization of these replication complexes. To address these limitations, we focus on Flock House virus (FHV), a nodavirus that infects Drosophila S2 cells, as a tractable model for RNA virus replication. Our previous results using classical thin-section TEM showed that protein A clusters mitochondria and induces membrane invagination [12,13], but only partial snapshots of the cell during infection were obtained, limiting our understanding of how these replication complexes assemble, expand, and interact with other cellular components throughout the infection cycle.
To overcome these limitations, we developed a Time-Snapshot Volume EM approach. In this method, Drosophila S2 cells are infected with FHV and sampled at multiple time points (e.g., 4 hours, 8 hours, and 17 hours post-infection) via high-pressure freezing (HPF). HPF can rapidly vitrify cells without ice crystal formation, preserving ultrastructural details in a near-native state. The resulting resin-embedded blocks are then sectioned into large consecutive series of thin sections, which can be collected onto coverslips for array tomography (see Figure 1). At each infection time point, large-area SEM images of these sections are recorded to build a complete volumetric reconstruction of infected S2 cells (see Figure 2). This approach allows us to track changes in mito-chondrial localization, the distribution of replication spherules, and any additional membrane rearrangements throughout the cell.
For more precise molecular identification, we will further perform immunofluorescent labeling on the same serial sections. We will target both FHV proteins, including the replicase protein A, the capsid protein, and the protein B1/B2 and host proteins, in-cluding mitochondrial membrane proteins. By overlaying fluorescence signals on the electron micrographs, we can correlate the two datasets to investigate how these viral factors localize relative to each other and to host proteins and organelles.
By comparing early and late time points, we aim to investigate how the localization of mitochondria is controlled during in-fection and how virus-host interactions change when virion assembly begins. Beyond FHV, the workflow described here can be extended to the study of other (+)RNA viruses, such as alphaviruses and flaviviruses, which shares similarities in host mem-brane remodeling for their genome replication.
In conclusion, Time-Snapshot Volume EM offers a powerful combination of high-resolution volumetric imaging and molecular specificity, enabling detailed visualization of the formation and progression of virus-induced replication compartments. By coup-ling HPF with large-scale array tomography reconstructions, we can investigate the spatial and temporal dynamics of replication complexes within entire infected cells.
Image ozaf048.382 figure 1
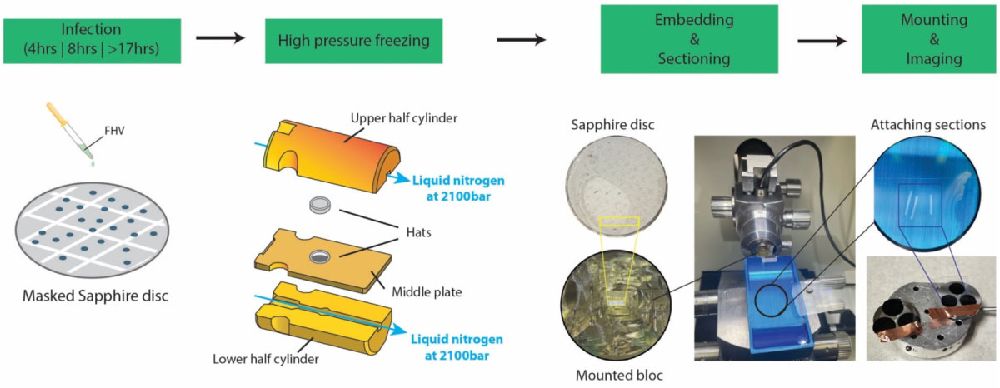
Fig. 1. Workflow for Volume EM analysis of FHV-infected S2 cells. S2 cells infected with FHV were seeded on carbon-coated sapphire discs and high-pressure frozen at multiple time points (4, 8, and >17 hours post-infection). Samples were then embedded in HM20 resin using an automatic freeze-substitution system. Ultrathin sections (120 nm) were cut with a Diatome Jumbo knife and collected on coverslips. Finally, images were acquired with a Zeiss Gemini 450 SEM.
Image ozaf048382 - figure 2
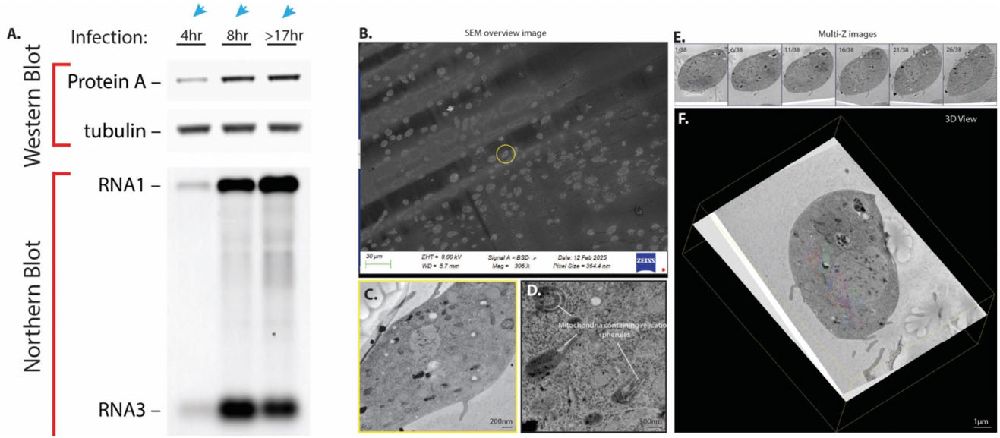
Fig. 2. 3D large-volume EM analysis of FHV-infected S2 cells at 4hr, 8hr, and >17hr post-infection. (A) Western blot showing FHV protein A expression levels and Northern blot detecting FHV genomic RNA1 and RNA3 across the three time points. (B) Overview of serial sections imaged by SEM. (C) SEM image (backscatter detector) of a 4hr-infected S2 cell. (D) High-magnification image revealing mitochondria-associated replication spherules in a 4hr-infected S2 cell. Scale bars = 200nm (left) and 500nm (right). (E) Multiple Z-planes from the serial-section dataset of a 4hr-infected S2 cell. (F) 3D reconstruction and mitochondrial segmentation of an intact 4hr-infected S2 cell. Scale bar = 1µm.
References
- Gustafsson MGL. Proceedings of the National Academy of Sciences. (2005) 102 13081–13086
- Betzig E et al. Science (2006) 313 1642–1645.
- Rust MJ et al. Nature Methods (2006) 3 793–796.
- Hein B et al. Proceedings of the National Academy of Sciences of the United States of America (2008) 105 14271–14276
- Schlegel P et al., Nature (2024) 634 139–152.
- Micheva KD and Smith SJ. Neuron (2007) 55 25–36.
- K. D. Micheva, N. O’Rourke, B. Busse, S. J. Smith, Cold Spring Harbor Protocols, in press, doi:10.1101/pdb.top89.
- Micheva KD et al. Cold Spring Harbor Protocols, in press. doi:10.1101/pdb.prot5525.
- Micheva KD et al. Cold Spring Harbor Protocols, in press, doi:10.1101/pdb.prot5524.
- Micheva KD et al. Cold Spring Harbor Protocols, in press, doi:10.1101/pdb.prot5526.
- Micheva KD and Bruchez MP. Current Opinion in Neurobiology (2012) 22 94–100.
- Kopek BG et al. Journal of Virology (2010) 84 12492–12503.
- Kopek BG et al. PLoS Biology (2007) 5 e220.
DMP-10 as Accelerators in Epoxy Resin Embedding for TEM Sample Preparation
Han Chen1,*
1TEM Core Facility, Department of Research Resource, The Pennsylvania State University, College of Medicine, Hershey, PA, USA
*Corresponding author: [email protected]
Effective TEM embedding requires uniform resin composition, good solubility, low viscosity, minimal shrinkage, beam stability, and excellent sectioning. However, achieving all these properties is challenging. Lower-viscosity resins often shrink significantly during polymerization, affecting sample integrity.
Since their introduction in 1956, epoxide resins have been widely used in TEM. Araldites offer superior sectioning but are high-ly viscous, while Epon 812 [1] improved handling with lower viscosity. Spurr resin [2] became popular for its penetration prop-erties, though it is less effective in aqueous uranyl acetate staining and has safety concerns.
Accelerators are essential for epoxy polymerization. DMP-30 and BDMA efficiently polymerize Araldite and Epon resins [3]. DMP-10, a dimethylaminomethyl phenol blend, increases viscosity and slows polymerization at room temperature, allowing bet-ter control. Its steric hindrance delays curing at lower temperatures, making it ideal for delicate biological samples.
This study utilized LX112 resin (Ladd Research) with DMP-10 (Tousimis) to embed mouse liver, heart, and skeletal muscle. Thin sections (70 nm) were cut with a Diatome 350 Ultra diamond knife on a Leica UC7 ultramicrotome, stained with uranyl acetate and lead citrate, and imaged using a JEOL JEM1400 TEM at Penn State College of Medicine TEM Facility (RRID: SCR_021200). The NANOSPRINT43M-MARKII camera (AMT) captured high-resolution images, revealing intact nuclear membranes, mitochondria with distinct cristae, and well-preserved cellular morphology with minimal distortion.
Our findings suggest LX112 resin with DMP-10 is a viable alternative to Spurr resin for muscle tissue embedding. Its slower polymerization rate enhances infiltration while maintaining optimal viscosity, making it a promising choice for TEM sample preparation, particularly for fragile biological specimens [4,5].
Image Ozaf048.423 figure 1
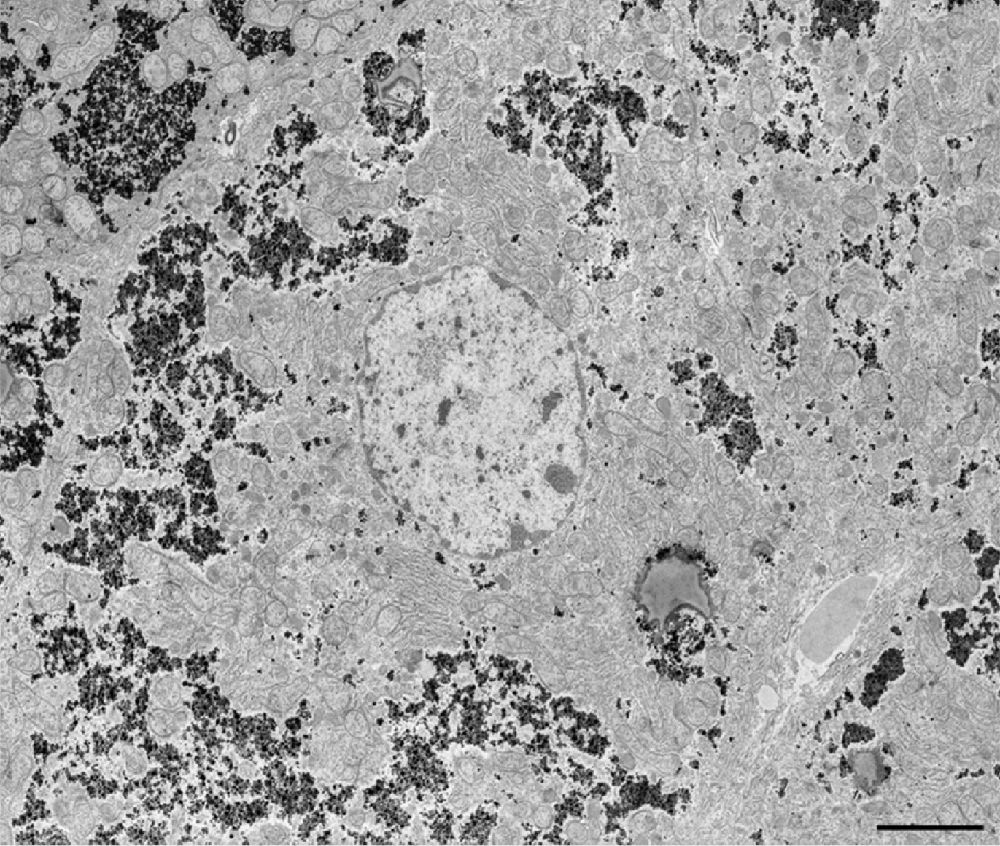
Fig. 1. Low magnification image of a representative field of the mouse liver. Scale bar = 4 um.
Image Ozaf048.423 figure 2
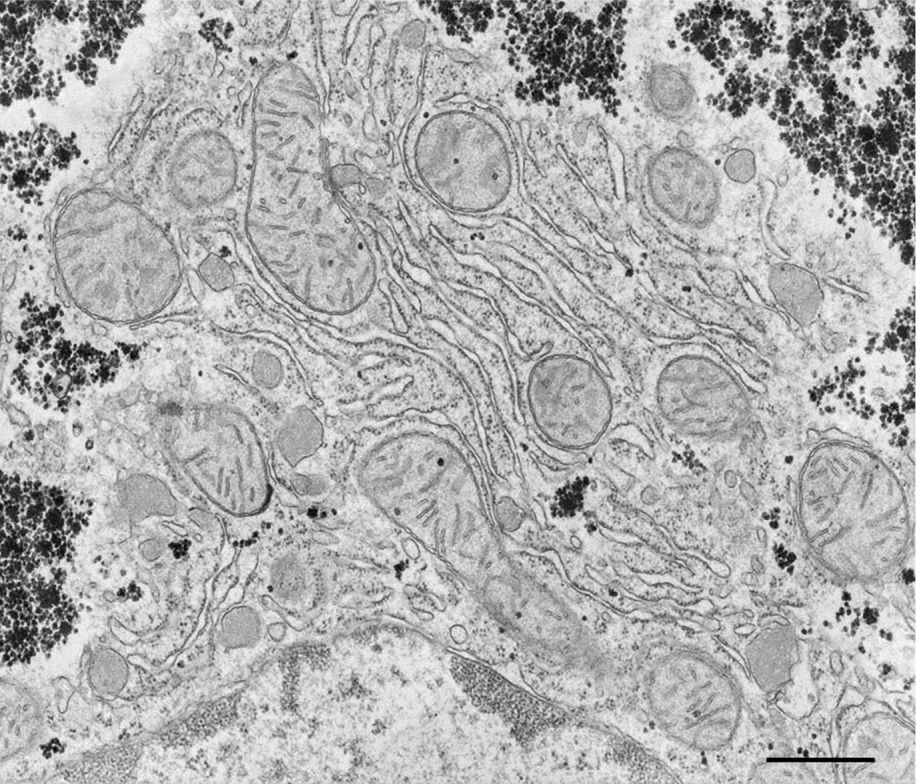
Fig. 2. Higher magnification image of mouse liver cell showing subcellular structures. Scale bar = 800 nm.
References
- Luft JH. Biophys. Biochem. Cytol. (1961) 9, 409.
- Spurr AR. Ultrastruct. Res. (1969) 26, 31.
- Glauert AM. Fixation, dehydration and embedding of biological. New York, NY: Elsevier North-Holland; 1975.
- The author acknowledges supports from the Penn State College of Medicine, Dr. H.G. Wang, and Dr. Ronggui Chen provide the mouse liver, heart, and skeletal muscle tissue.
- I declare that I have no personal or financial interest in Ledd Research and Tousimis and my findings in this paper are solely based on objective research.


